|
|
Лабораторное занятие № 3. Строение и функции биополимеров. Выделение и количественное определение биополимеров различными методами.
Известно, что все организмы состоят, из клеток и продукта их жизнедеятельности = внеклеточного вещества. Причем прокариоты, простейшие и грибы, встречаются в виде отдельных клеток или их колоний. Многоклеточные же эукариоты, типа высших растений и животных, обычно состоят из множеств клеточных дифферонов, структурно сопряженных друг с другом в ткани, согласованные в своем метаболизме и жизненных циклах в интересах организмов, их популяций и сообществ. Залог успеха разрушающего биохимического анализа – рациональный выбор биообъекта, щадящие и верные приемы изъятия из организма образца ткани, его фиксации, хранения и обработки. Различия же в целях и методах анализа (рис. 1) проявились к началу ХХ в.
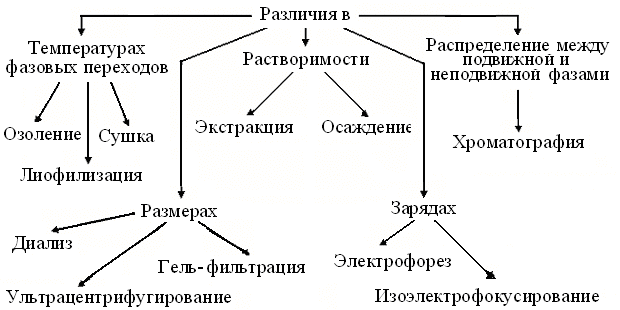
Рис. 1. Схема целей и этапов биохимических исследований
Изучение структуры и функций биомолекул потребовало их выделения и очистки до минимума примесей, а в идеале – до однородного состояния. Но быстрота разрушения части молекул, например, с функцией медиаторов, как и высокая вероятность денатурации белков, часто вынуждают хранить и вести выделение препаратовна холоде. Для извлечения = экстракции большинства белков из клеток животных, обычно достаточно механической гомогенизации в водной среде выделения. Разрушение же клеточных стенок растений и, особенно микроорганизмов, требует шаровых мельниц, ультразвука и других жестких приемов. Удаление из гомогенатов обрывков волокон, фрагментов клеточных структур и т.п., ведут с помощью их отстаивания, фильтрации или центрифугирования, а для удаления или замены низкомолекулярных веществ, пользуются диализом,ультрафильтрацией или молекулярными ситами. Трудоемкость процедур препаративной биохимии и обилие специфических терминов поясняет схема (рис.2).
Рис. 2. Схема методов препаративной биохимии
Очевидно, что процесс выделения и очистки препарата почти неизбежно связан с его разбавлением, требующим потом, адекватного задаче концентрирования раствора. Часто для этого применяют высаливание белков, то есть их осаждение из раствора с помощью больших концентраций нейтральных солей: NaCl, (NH4)2SO4, MgSO4 и т.д., в соответствии с положением ионов в ряду Гофмейстера. Этот прием основан на: 1) нейтрализации зарядов молекул белка с помощью противоионов и 2) обратимом разрушении их гидратных оболочек. Т.к. высаливание зависит от размеров и гидрофобности макромолекул, то глобулины плазмы крови оседают при 50 % концентрации сульфата аммония, а осаждение сывороточного альбумина = СА, требует его 100 % насыщения. Т.о., высаливание позволяет не только концентрировать целевой продукт, но и обеспечивает его дополнительную очистку. Осадки белков после этого, растворяют добавлением воды или повторным диализом, добиваясь 50-80 % концентрации препарата. Такой продукт называют сырцом и подвергают заключительным этапам очистки и проверки гомогенности, с помощью разных вариантов хроматографии и электрофореза. Для диагностики состояний животных и людей, физиологи и врачи неоднократно пытались применять доступные покровные ткани, секреты и экскреты, но быстро убеждались в их ограниченной информативности. Поэтому, уже свыше 100 лет, основным источником информации служит химический анализ слюны, желудочного сока, желчи, молока и, особенно крови, получаемых с помощью малоболезненных и относительно простых и безопасных процедур. Понятно, что точность основных аналитических процедур, уже рассмотренных на предыдущих занятиях, зависит от правильного взятия проб, например, крови – натощак и т.д. и точного выполнения приемов хранения и подготовки материалов к анализу, в основном совпадающих с вышеописанными методами препаративной биохимии. К сожалению, большинство диагностических лабораторий РФ, как и 100 лет назад, ведет эти анализы вручную, трудоемкими методами микроскопии и классической биохимии. Поэтому, относительно точные = эталонные результаты диагностических показателей выдаются через часы или сутки, а на их получение, идет свыше трети трудозатрат соответствующих лабораторий. Тем не менее, именно они дают от 80 до 100 % объективной и полезной информации о молекулярно-клеточном состоянии и системах управления внутренней средой организма. Основы спектроскопии
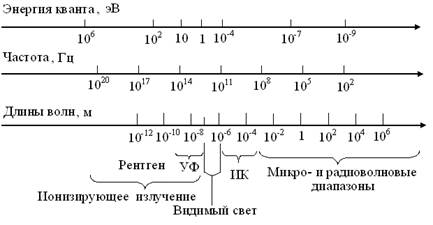
Известно, что видимый свет, лишь малая часть спектра электромагнитных излучений, чьи взаимно перпендикулярные электрическая и магнитная компоненты, периодически меняются местами. Поэтому энергию кванта (ε), частоту (ν) и длину их волн (λ) объединяют жесткие зависимости (рис. 3). В соответствии с ними, электромагнитный спектр условно делят на частично перекрывающиеся диапазоны, отчасти зависящие от источника излучений. Так, нет принципиальной разницы между волнами одной и той же длины в ИК и микроволновом диапазонах. Но, их называют ИК-лучам при генерации тепловым прибором и относят к микроволнам, если он электронный.
Рис. 3. Схема шкал электромагнитного спектра
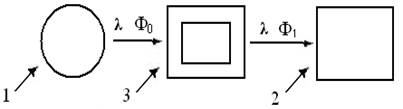
Из блок-схемы (рис. 4) видно, что независимо от диапазона, все спектрометры включают, т. н. оптопару: 1. источник монохромного излучения энергии с длиной волны X и 2. его приемник = детектор, преобразующий сигнал в электроток индикатора.
Рис. 4. Принципиальная схема однолучевых спектрометров
Третью составляющую – кювету с изучаемым образцом (3) размещают между ними, сопоставляя потоки энергии излучателя Фо и детектора Ф1, для получения информации о структуре и свойствах объекта исследований. Так удалось выделить 2 класса спектров: эмиссии = испускания и абсорбции = поглощения. Известно, что по закону Г. Кирхгофа (1859) – любое вещество хорошопоглощает излучения именно тех λ,которые само испускает. В свою очередь, каждый класс излучений подразделяют на непрерывные = сплошные, полосатые и линейчатые спектры. Эмпирические данные И. Ньютон (1666), Й. Фраунгофер (1814) и др. и закон взаимосвязи спектров поглощения и испускания, позволили Г. Кирхгофу и Р.Бунзену открыть важнейший закон физической оптики: каждый атом (функциональная группа, молекула, кристалл) испускает характерный линейчатый спектр электромагнитных волн. На этой основе была создана теория характеристических спектров, позволившая открыть He, Rb, и др. химические элементы, а в ХХ в. – сделать приборы и методы спектрального анализа универсальным инструментом изучения законов существования Вселенной (табл. 1). Таблица 1 Характеристика основных диапазонов электромагнитного спектра
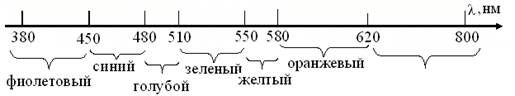
Применительно к химии и биологии важно, что энергия квантов излучений с λ < 1 нм, разрушает атомные ядра и химические связи, тогда как у волн длиной > 1000 нм, недостаточно энергии для изменений молекулярной структуры вещества. Поэтому основой идентификации веществ стали ИК и, реже УФ диапазоны. А в основу фоторецепции клеток и организмов, лег эволюционный отбор молекул красящих и зрительных фотопигментов, способных при поглощении квантов излучений промежуточной области, максимально изменять орбитали электронов, участвующих в образовании сопряженных двойных связей. Как сказано выше, понятие свет относят к области электромагнитных волн в диапазоне 10-8-10-6 м (рис.5). Человек и другие млекопитающие видят лишь в небольшой части спектра, соответственно длинам волн 380 – 800 нм. Но, люди с удаленным хрусталиком, воспринимают УФ-диапазон, как и насекомые, которых привлекают, невзрачные для млекопитающих цветы. При том известно, что птицы – не видят в синей области спектра, ряд пресмыкающихся воспринимает ИК лучи, а летучие мыши и дельфины пользуются ультразвуковой эхолокацией.
Рис. 5. Видимая область спектра для млекопитающих
Фотометрический анализ – совокупность методов качественного и количественного анализа по интенсивности УФ, видимого и ИК излучений. Но, т.к. биообъекты на 2/3 состоят из воды, высокая теплоемкость которой поглощает ИК лучи, то в биологии их почти не применяют, широко используя 2 других диапазона (рис.6).
Рис.6. Схема методов фотометрии ( по http://www.krugosvet.ru/ articles/ 23/ 1002315/1002315a1.htm)
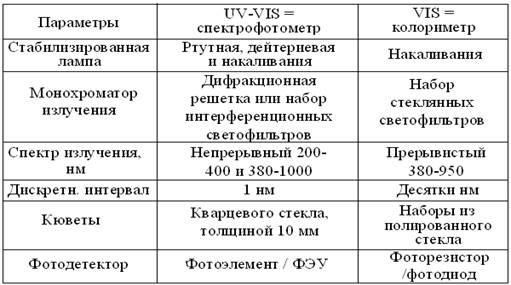
Нередко фотометрию сводят к молекулярно-абсорбционному анализу, основанному на объединенном законе светопоглощения Бугера-Ламберта-Бера. Согласно ему, в стандартных условиях кюветного отделения и длины волны, перпендикулярной толщине слоя однородной среды (l), между концентрацией (С) поглощающего вещества и соотношением интенсивностей исходного = Ф0 и достигшего детектора = Ф1 потоков излучений, возникает логарифмическая зависимость: С = lg Ф0/Ф1. Отсюда появился не очень точный, но распространенный синоним – концентрационный анализ. При стандартных λ и толщине образца, количество света достигшее фотодетектора выражают линейной шкалой светопропускания = Т (transition), которую для удобства в работе, обычно градуируют в %. К сожалению, она не имеет прямого отношения к свойствам образца, ослабляющего интенсивность излучений за счет поглощения = А (absorbtion) или, таких устаревших понятий, как оптическая плотность = D (density) раствора и коэффициент его экстинкции = Е. С другой стороны, зависимость 2 – lg T = А, обеспечивает высокую точность измерения концентраций в диапазоне 0,2-0,8 безразмерной логарифмической шкалы от 0 до ∞. Во избежание оптических эффектов, способных исказить результаты, фотометрию проводят в закрытых кюветных отделениях фотометров, а хрупкие кюветы с плоскопараллельными стенкам – берегут от падений, царапин, потеков и капель. По той же причине, нельзя касаться руками оптических стенок кювет, а после употребления – их тщательно моют с мылом и хранят в дистилляте, высушив перед следующим измерением. Чтоб кюветы не запотевали, их заполняют растворами комнатной температуры до уровня, при котором поток света целиком проходит через раствор. Таблица 2 Сравнительная характеристика фотометров
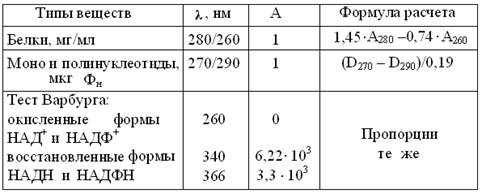
В таблице 2 представлены важнейшие эксплуатационные характеристики однолучевых фотометров, предназначенных для измерений в УФ и видимой областях спектра. Так как у колориметров они гораздо проще, то и стоимость их ниже, примерно на порядок. Спектрофотометрия в УФ диапазоне позволяет: 1) идентифицировать вещества по их характерным спектрам поглощения; 2) при стандартной длине УФ лучей измерять в 1 М водных растворах многих веществ, их важнейшую спектральную характеристику – молярный коэффициент абсорбции (табл. 3). Этот неразрушающий метод позволяет экономить препарат, сводя измерение концентраций к замеру спектральных максимумов поглощения биомолекул. Аналогично, рутинные измерения активности ферментов упрощают до т.н. теста Варбурга, определяя в стандартных условиях, прирост или убыль 1 мкмоль НАДН (НАДФН). Так как большинство веществ поглощает видимый свет незначительно, то окрашенные продукты, в количестве, пропорциональном концентрации исходного вещества, получают из них после определенных физико-химических воздействий, включая качественные реакции. Этот прием широко применяют для колориметрии в относительно простых фотоэлектроколориметрах = ФЭКах. Из биомолекул, так определяют концентрации ортофосфата = неорганический фосфат (Фн) и других фосфоорганических соединений, железа, мочевины, билирубина, холестерола, жирных кислот, белка, гемоглобина и др. До недавних пор, мирясь с недостатками, так же определяли глюкозу, фруктозу, крахмал, гликоген и др. сахара, пока колориметрию углеводов не вытеснили ферментные и электрометрические метода анализа. Таблица 3 Расчет концентраций биомолекул по их спектральным характеристикам УФО
При колориметрии, изучаемый раствор сравнивают с поглощением: 1. контрольного образца и 2. проб стандартного ряда = шкалы сравнений или графика калибровки. В качестве контроля обычно используют растворитель или раствор со всеми компонентами, кроме исследуемого вещества. Напротив, чтобы концентрация анализируемого вещества пропорционально убывала в 3 – 5 параллельных пробах шкалы сравнений, ее готовят разведением раствора точной концентрации, с учетом чувствительности конкретного метода, температуры среды и времени развития окраски. Результаты колориметрии проб шкалы сравнений оформляют в виде графика зависимости интенсивности окраски (ось ординат) от концентрации анализируемого вещества (ось абсцисс), добиваясь его прямолинейности. За редкими исключениями, полученный график нельзя экстраполировать за пределы экспериментальных точек. Оптимизируя размер кювет нужно стремиться, чтобы значения А калибровочного графика уложились в интервале 0,1 – 1,0, а еще лучше – 0,1 – 0,6. Так как все ФЭКи различаются степенью монохроматизации света и, соответственно, чувствительностью, недопустимо пользоваться калибровочным графиком или таблицей соотношений А и С, построенными для других ФЭКов! Больше того, калибровки, построенные с реактивами разных партий, как правило, не совпадают. Поэтому при смене реактивов, график приходится строить заново. После измерений абсорбции в опытных пробах, концентрации анализируемого вещества в них определяют в соответствии с калибровочным графиком или таблицей, с учетом объема исследуемого раствора. Если величина А пробы окажется выше имеющихся значений графика (таблицы), во избежание искажения результатов, разбавлять реакционную смесь для повторной колориметрии не рекомендуется. В этих случаях, лучше повторить колориметрический тест с меньшим объемом исследуемого материала, чтоб его результаты попали на калибровку. Дополнительная литература
3. Бурштейн Э. А. Люминесценция белковых хромофоров. – М.: ВИНИТИ «Итоги науки и техники». Серия БИОФИЗИКА. – Т. 6, 1976. – 216 с. 4. Черницкий Е. А. Люминесценция и структурная лабильность белков в растворе и клетке. – Минск.: Наука и техника, 1972. – 280 с.
18. Троицкий Г. В. Дефектные белки. Постсинтетическая модификация. – Киев.: Наукова думка, 1991. – 232 с.
2. Инструкция по эксплуатации фотоэлектроколориметра = ФЭКа, модели КФК-2 1. За 15 мин. до начала фотометрии, под контролем лаборанта или преподавателя, дежурный убеждается, что стрелка гальванометра находится в положении «механический 0», открывает крышку кюветного отделения прибора, вставляет его вилку в сетевую розетку и, тумблером на задней панели слева, включает прогрев ФЭКа. 2. Перед началом измерений, вращая рукоять светофильтра, на передней панели прибора слева, установить нужный диапазон светового потока = пучка. 3. Убедиться, что рукоять чувствительности прибора на передней панели справа, соответствует цветовому диапазону рукояти светофильтра. 4. Подготовить фотометрические кюветы нужной толщины, насухо протереть марлевой салфеткой их оптические грани и, убедиться в отсутствии на них пятен и др. грязи. ВНИМАНИЕ! Поскольку каждый ФЭК оснащен лишь одним набором кювет, изготовленных из полированного оптического стекла, пользоваться ими можно лишь над рабочим столом и брать рукой, только за боковые грани, оберегая от царапин и ударов. 5. Убедиться, что стрелка включенного гальванометра находится в положении «электрический 0». 6. Заполнить контрольную кювету дистиллятом или, указанным в соответствующей методике контрольным раствором, убедиться в отсутствии капель и потеков на ее оптических гранях, строго вертикально вставить ее в дальнее гнездо кюветодержателя и, рукояткой в центре передней панели ФЭКа, до упора ввести кювету в световой пучок. 7. В соответствии с п. 6, заполнить вторую кювету исследуемым раствором и вставить ее в свободное гнездо кюветодержателя, примерно на том же расстоянии от источника света. ВНИМАНИЕ! При анализе серии однотипных образцов, избегая излишних промывок кюветы, начинать фотометрию с наиболее разбавленных проб. 8. Закрыть крышку кюветного отделения до звяка шторки и, вращая на передней панели вверху рукояти грубой, а затем и тонкой настройки, установить стрелку гальванометра на 0 шкалы D = А. ВНИМАНИЕ! При положении стрелки между делениями шкалы - округлять цифру до целого значения. 9. Не открывая крышки кюветного отделения, переместить до упора рукоять кюветодержателя в противоположение, заменив тем самым в световом потоке, контрольную кювету исследуемой. 10. Убедиться, что стрелка гальванометра успокоилась и занести показания ФЭКа в протокол опыта. 11. Открыть крышку кюветного отделения, вынуть из кюветодержателя кювету с исследуемым раствором и, декантировать ее содержимое обратно в соответствующую пробирку. ВНИМАНИЕ! Для последующих измерений, во избежание потеков на стенках, кювету не переворачивать, а удерживая вверх дном, осушить ее торец фильтровальной бумагой. 12. При последующих измерениях, руководствоваться пп. 6-11 данной инструкции. Экспериментальная часть Проводится в соответствии со схемой на рис. 7 (делает только дежурная пара)
Рис.7. Схема деления крови на фракции (пояснения в тексте)
|
|